Neurodegenerative conditions such as Parkinson’s and Alzheimer’s diseases constitute a major human-health burden. Although the symptoms, or the cells affected, can differ in such disorders, some neurodegenerative diseases have certain characteristics in common. These include a state of inflammation and impaired elimination of defective mitochondrial organelles. However, it remains to be determined whether such common alterations are interconnected, and whether they are a cause or a consequence of disease. Writing in Nature, Sliter et al. report their investigation of mice that have alterations in genes linked to Parkinson’s disease. The authors identify a direct connection between the cellular process that eliminates damaged mitochondria called mitophagy and inflammation.
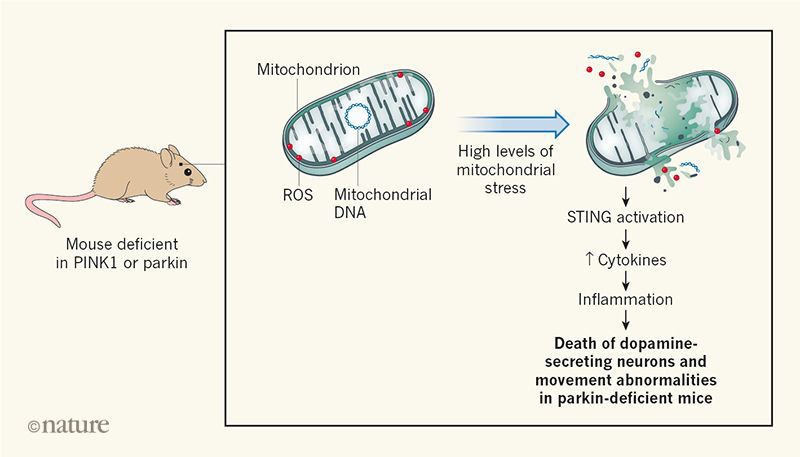
The enzymes PINK1 and parkin act in a pathway that attaches a protein called ubiquitin to cellular proteins; such ubiquitin-tagged components are targeted for cellular destruction. These enzymes assist with the process of mitophagy, in which non-functional mitochondrial fragments are rapidly sequestered into a membrane-bound vesicle that is degraded when it fuses with an organelle known as a lysosome.
Mutations that prevent the normal expression of
PINK1 or parkin are linked to an early-onset form of Parkinson’s disease, and there is evidence that failure to successfully eliminate damaged mitochondria results in a higher risk of developing the disease. However, mice that are deficient in PINK1 or parkin do not develop symptoms of the type observed in people who have abnormalities in the expression of these proteins; such symptoms include movement problems arising from the loss of neuronal cells that produce the neurotransmitter molecule dopamine. Nor do these animals have the high level of inflammation that is a hallmark of Parkinson’s disease.
The finding that the loss of
PINK1 or parkin has a minimal effect on animals was surprising, because it was long thought that the removal of damaged mitochondria serves a key role in protecting cells from oxidative damage. Defective mitochondria represent a severe threat to cells because ruptured mitochondria might release reactive oxygen species (ROS) that cause substantial cellular damage. For example, ROS might increase the burden of potentially toxic protein aggregates if proteins are subject to ROS-mediated damage. Defective mitochondria might also release components that are not normally present in the cytoplasm, such as mitochondrial DNA. Indeed, the intrusion of mitochondrial DNA into the cytoplasm can trigger inflammation mediated by the protein STING. This raises the question of whether protection from inflammation, rather than from oxidative damage, might be the key role of mitophagy in the context of Parkinson’s disease.
Sliter et al. investigated the consequences of
PINK1 or parkin loss in mice that were subjected to a high level of mitochondrial stress. This stress was produced either by subjecting animals to an intensive, exhausting exercise regime or by exploiting a genetic alteration found in animals termed mutator mice in which a defective polymerase enzyme causes a high level of mitochondrial-DNA mutations. It was previously reported that old mutator mice that lack parkin have fewer dopamine-secreting neurons than normal, and that these mice develop movement abnormalities that are reminiscent of those observed in people who have Parkinson’s disease. When the authors imposed mitochondrial stress on animals lacking PINK1 or parkin, they found that the bloodstream level of inflammation-driving molecules called cytokines was much higher than it was in mice that were not subjected to this mitochondrial stress.
However, the authors found that if mice lacked STING, as well as PINK1 or parkin, the expression of inflammatory cytokines did not increase as a result of mitochondrial stress. This indicated that STING is required to drive the inflammation mediated by this type of stress. Moreover, an absence of STING prevented the movement defects and neuronal losses that usually occur in old mutator mice that lack parkin. The authors found that the bloodstream levels of the inflammatory cytokines
IL-6,
IL-1β and
CCL2, which are elevated above normal in old mutator mice that lack parkin, are also higher than normal in people with Parkinson’s disease who have mutations in both copies of the parkin gene. However, the authors observed that these cytokines were also elevated in disease-free relatives of people who have Parkinson’s disease. Sliter and colleagues’ study of these relatives, who have a mutation in only one of their two copies of the parkin gene, suggests that these particular cytokine alterations are not sufficient to cause the disease. Interestingly, people who receive long-term treatment with non-steroidal anti-inflammatory drugs have a lower than average risk of developing Parkinson’s disease. This observation is consistent with a model in which low levels of inflammation might protect gainst neurodegeneration.
Sherry
 Phone:
Phone:  E-mail:
E-mail:  Address:
Address: